
開篇宣告:1:以下內容轉自以下內容轉自網路,如有侵權,可聯絡刪除
2. 以下觀點,僅代表作者個人觀點,請帶著質疑的態度去閱讀。如有問題,歡迎評論區留言或直接聯絡作者
撰稿人:秦奕菲 沙琳琳
審閱人:袁鷹 葛妙妙 郭文天
摘要
2022年2月25日,DIA中國統計論壇第十期——FDA對BOIN方法的稽覈及修改意見解析——順利召開。本次講座特邀中國醫學科學院腫瘤醫院肝膽外科副主任趙宏教授擔任主持,由思特爾(Cytel)中國公司高階諮詢師秦奕菲博士主講,特邀美國MD Anderson 癌症中心教授、BOIN設計原創發明人袁鷹教授,勃林格殷格翰製藥公司(BI, US)腫瘤領域專家統計師葛妙妙博士,恆瑞醫藥臨床研發部統計助理總監郭文天博士作為討論嘉賓,詳細介紹了FDA對BOIN方法的稽覈及修改意見,並針對大家關注的BOIN方法的焦點問題展開討論。
背景
BOIN設計,全稱是貝葉斯最優區間設計(Bayesian Optimal Interval design),是由美國德克薩斯大學MD Anderson癌症中心的袁鷹教授團隊在2015年提出的一種I期臨床試驗劑量探索設計方法,具有操作簡便,效能優良的特點。2021年12月10日,BOIN設計獲得美國FDA Fit-for-Purpose官宣認證,肯定了BOIN設計的優越效能,並認證其適用於研究目的是確定最大耐受劑量(MTD)的I期臨床試驗,也是繼獲得中國CDE認可後(詳見《抗腫瘤藥物臨床試驗統計學設計指導原則(試行)》),BOIN設計在全球重要藥品監管機構取得的又一重要認證。本期論壇重磅邀請BOIN設計方法學創始團隊、國內外企業統計師代表及資深臨床研究者,共同分享BOIN設計的理念與方法、探討和解析FDA對BOIN方法的稽覈及意見,為I期劑量遞增適宜方法選擇提供參考。
關於Fit-for-Purpose (FFP) 認證
FFP認證是FDA針對藥物開發工具(DDT)的一項認證。獲得FFP認證意味著該DDT已預先透過FDA的稽覈。採用FFP認證的DDT進行藥物開發活動,可以節省與FDA溝通的時間。此次BOIN設計是作為I期劑量探索的統計學方法獲得FFP認證。
關於BOIN設計
BOIN設計是由Liu&Yuan(2015)提出的I期劑量探索方法。其基本原理和實現過程如下:
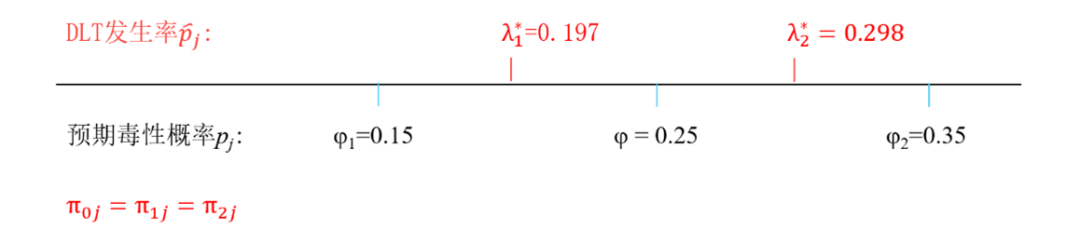
1. 設定一個目標毒性機率φ,一個較低毒性機率φ1和一個較高毒性機率φ2,適用於所有劑量。這三個引數要結合研究藥物情況與臨床研究者共同討論確定。本文假設φ=0.25,φ1=0.15,φ2=0.35。
2. 在貝葉斯框架下,為φ,φ1,φ2各分配一個先驗機率,即預先估計φ,φ1,φ2發生的可能性大小。此次FFP認證的是無資訊先驗下的BOIN方法,即φ,φ1,φ2發生的可能性相等,各為1/3。
3. 爲了使決策錯誤最小化,先定義正確的決策和錯誤的決策,p_j在劑量j的條件下正確的毒性機率.H_0j是現在的劑量d_j就是MTD,決策應為維持。H_1j是現在的劑量d_j低於MTD,決策應為上升。H_2j是現在的劑量d_j高於MTD,決策應為下降。若做出正確的決策為R,ℇ,D,對應錯誤的決策為R ̅,ℇ ̅,D ̅。利用φ,φ1,φ2的先驗機率,建立決策錯誤率(Decision error)函式。
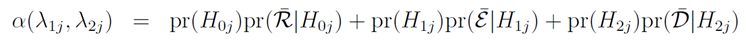
求解極小值,在無資訊先驗下可以獲得一個特例解λ1*和λ2*:
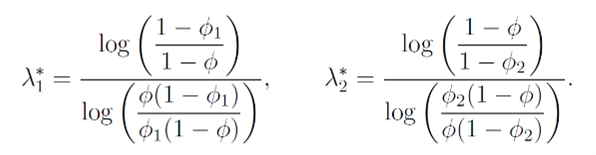
當觀測值(p_j ) ̂≤ λ_1^*時,可以上升一個劑量,因為在這個條件下,當前劑量還未達到MTD。觀測值(p_j ) ̂> λ_2^*時,可以下降一個劑量,因為在這個條件下,當前劑量已經超過MTD。這個解適用於所有劑量水平並且不受組樣本量(cohort size)的影響。
具體的尋找合適劑量流程圖圖下圖所示。
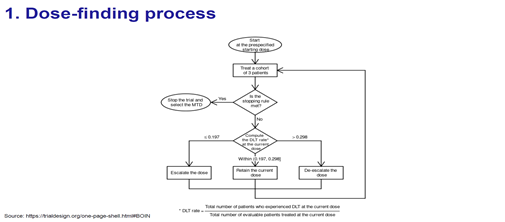
4. 根據修改後的定理1,λ1*和λ2*可以作為判斷劑量上升/下降/保持的兩個界值點。
透過比較DLT發生率與 λ1*和λ2*的大小,判斷劑量上升/下降/保持,可以使決策錯誤率最小。
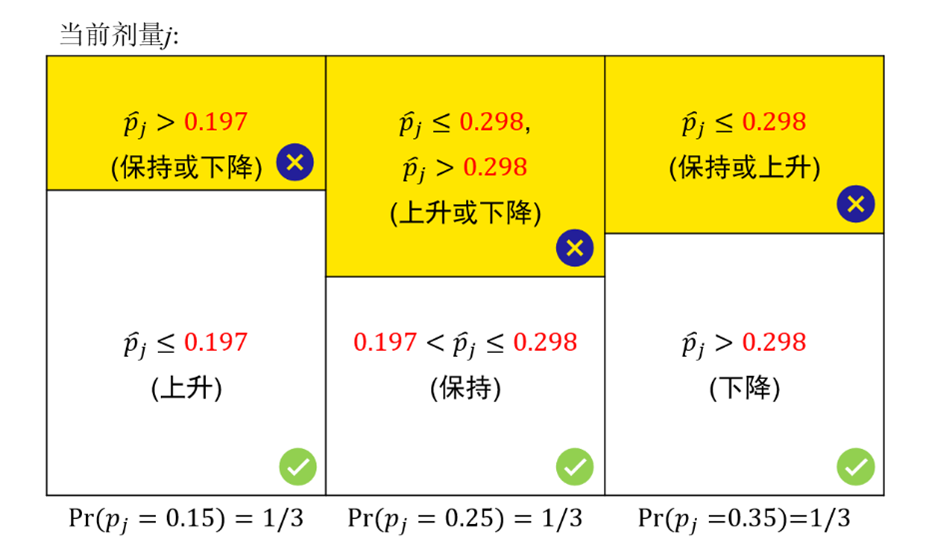
5. 在所有劑量上重複上述過程,直至達到試驗終止條件。
FDA的稽覈意見
FDA在稽覈Liu and Yuan (2015) 時,發現了一些technical issues。申請人根據FDA的意見對相關問題做了改進。Fit-for-Purpose認證是針對改進後的BOIN設計。
申請人提交的材料,包括模擬研究,都是針對無資訊先驗下的local BOIN設計,因此Fit-for-Purpose認證也是僅針對無資訊先驗下的local BOIN設計。
申請人針對若干種I期劑量探索方法進行了模擬研究和對比。BOIN generally performed well in the simulation scenarios considered(但也不是說其他方法的表現不好)。在某些情況下,例如:劑量毒性關係非單調遞增,或者存在毒性延遲,BOIN方法可能不適用。
結論:A multidisciplinary team … has reviewed all aspects of the submission. The review team finds that under the non-informative prior, the local BOIN design, in its revised form, can be designated fit-for-purpose.
授予BOIN方法FFP認證,並不排除其他I期劑量探索方法的使用。也不排除BOIN方法本身在非local的情況和有資訊先驗情況下的使用。
建議申請人的相關軟體與BOIN方法的改進保持一致。
BOIN方法的修改及影響
1. 調整劑量上升/下降規則
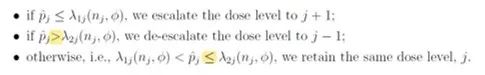
原規則為DLT發生率大於等於λ2j時,降低劑量。現修改爲DLT發生率大於λ2j時,降低劑量。原規則為DLT發生率大於λ1j,小於λ2j時,保持劑量。現修改爲DLT發生率大於λ1j,小於等於λ2j時,保持劑量。
由於現實中DLT發生率幾乎不可能等於λ2j,此修改對於BOIN方法的使用沒有影響。
2. 將界值點擴充套件為界值區間
假設cohort size=3,則DLT發生率為0/3,1/3,2/3,3/3。FDA認為介於0和1/3之間的任何值均可以作為界值,而不僅限於λ1*=0.197和λ2*=0.298。
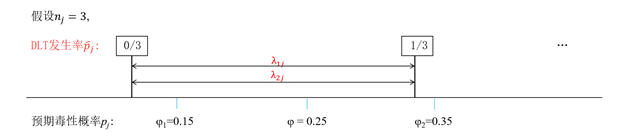
作者根據FDA的意見,將原文中的界值點(point solutions)擴充套件為界值區間(interval solutions)。修改後的解為區間形式。
由於原方法中的界值點λ1*和λ2*已包含在界值區間λ1j和λ2j中,是修改後的解的一個特例,所以此修改對於BOIN方法的使用沒有影響。
3. FFP認證的是無資訊先驗下的BOIN方法
由於在某些極端情況下,有資訊先驗的BOIN方法可能無法給出合理的λ1j和λ2j,例如:λ1j > λ2j,因此FDA只認證了無資訊先驗下的BOIN方法。作者相應修改了定理1,增加了無資訊先驗的限制條件。
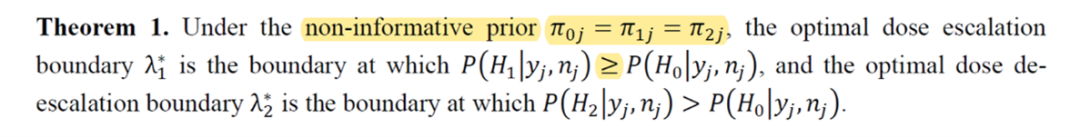
如果是在無資訊先驗下使用BOIN方法,則不受此修改的影響。
總結
FFP認證是針對無資訊先驗下的、改進後的Local BOIN方法(本文中介紹的方法即為Local BOIN方法)。
除了採用有資訊先驗的情況外,FDA對於BOIN方法的修改意見對於已採用BOIN方法的試驗設計沒有影響。如果採用有資訊先驗的BOIN設計,建議重新評估。
FDA並未排除有資訊先驗下BOIN方法的使用,只是需要注意避免可能存在的問題。同時,FDA也未排除其他非FFP認證的I期劑量探索方法。
現有軟體(如Trialdesign, EastBayes等)均採用無資訊先驗下的BOIN方法。如果是採用這些軟體進行BOIN設計,不受此次修改的影響。
嘉賓討論
討論1:目標毒性機率Φ、過低毒性機率Φ1和過高毒性機率Φ2的選擇
BOIN方法本身並不限制Φ1和Φ2的取值(只要在0~1之間). Φ1 represents a significantly underdosing toxicity rate such that the dose escalation is necessary; and Φ2 represents a significantly overdosing toxicity rate such that the dose escalation is necessary. BOIN minimizes the decision rule over these three cases. 因此,如果Φ1與Φ之間或Φ與Φ2之間的差異過小,在小樣本下無法有效分辨這種差異帶來的影響,optimization is not practically relevant。根據實踐經驗,Φ1=0.6Φ,Φ2=1.4Φ時,所獲得的劑量探索結果與臨床醫生的經驗判斷相吻合。通常對Φ1和Φ2不需要進行calibrate。如果希望更保守一些,可以降低Φ2。
在I期劑量探索過程中,對於毒性有客觀評價標準,例如:肝功能等血液指標,代謝指標等。這些客觀指標會體現在方案中,並根據這些指標評價用藥後的毒性。Φ、Φ1、Φ2一般由臨床醫生結合研究藥物的特性,以及臨床實踐經驗等進行選擇,再透過模擬進行校準。目前常用的目標毒性機率Φ是25~30%。
討論2:有資訊先驗與無資訊先驗
The effect of using informative prior is to influence the escalation and de-escalation boundary λ1* and λ2*。例如:如果給與Φ1較大的先驗機率 (i.e., the prior says that the dose is very safe),那麼當DLT發生率較高時,例如2/3,模型的決策可能仍然是保持甚至上升,但這通常與倫理和臨床實際操作不符。I期試驗中安全性是最重要的。The most important feature of dose finding is that we assess the safety of the dose sequentially one level at a time from low to high. 這就像摸著石頭過河,即使我們知道當前這一步是安全的,我們並不知道下一步是否依然安全。Each dose level has the chance to be safe, acceptable or toxic. Thus, the most natural approach is to set non-informative prior for each level. Letting the DLT rate dominate the decision is safe and can avoid the conflict that the data show the dose is toxic (e.g., 2/3), but we still stay or escalation the dose because the prior says that it is safe.
如果同一個研究藥物已在一個地區或人群上獲得資料,這個資訊可以應用於該藥物在另一地區或人群上的研究,此時可以考慮有資訊先驗,但在借用資訊時要格外謹慎。The prior information should not dominate the trial data. The historical information can be down weighted (e.g. through mixture prior distribution with non-informative component to control the effective sample size) to make sure that reasonable escalation decisions will be made when only limited data is observed in the trial, and that the calibrated informative prior can still improve the design efficiency. 先驗資訊也可以源自已上市同類藥物或臨床前資料,但這種資訊借鑑的可靠性較難把握。
考慮到先驗資訊的可靠性問題,建議將先驗資訊用於確定進入研究的各個劑量水平,而不一定要放在模型中參與劑量上升/下降決策。總之,先驗資訊可以輔助決策,但應避免dominating the decision。
討論3:界值點(point solutions)與界值區間(interval solutions)
將界值點修改爲界值區間對於BOIN方法的使用沒有任何影響。因為原方法中的解(λ1*和λ2*)就是位於界值區間內的一個特殊的解。在無資訊先驗下,界值點和界值區間做出的決策結果完全一致。此時,λ1*和λ2*是簡單的解析解,而且作為劑量上升/下降決策的標準,不論cohort size或dose level是多少,這個標準並不隨之改變,非常便於實際應用。
討論4:如何確定一次入組的受試者人數(cohort size)?
實踐中cohort size=3是沿用了傳統3+3的慣例。同時也有臨床的考量。如果cohort size等於1或2,會導致試驗週期過長。如果cohort size較大,可能使更多受試者暴露在無效或過毒的劑量上。所以cohort size=3是一個折衷的選擇。
在劑量很低的水平上,也可以進行加速滴定。注意此時不宜使用DLT作為毒性標準,應採用更低的毒性標準。如果確認有毒性發生,則應繼續在當前劑量入組病人,並使用cohort size=3(一般來說是這樣)的設計進行餘下的劑量爬坡試驗。
討論5:選擇無資訊先驗的BOIN設計和選擇多個先驗skeleton的BMA-CRM設計相比,哪個更具合理性?
BMA-CRM採用dose-toxicity 模型進行毒性估計和決策。Although it adopts multiple model and thus more robust than the CRM, it is still influenced by the model misspecification. 如果模型與真實情況相近,則效果好於無資訊先驗的BOIN設計。反之,則效果不好。BOIN does not make any assumption on dose-toxicity curve, thus more robust. 並且,BOIN設計採用無資訊先驗對於結果的影響不論好壞都比較有限,起作用的主要是實際觀察到的DLT發生率。
討論6:可否同時開展dose N和dose N+1試驗?
可以。但當決策結果出現衝突時,例如,劑量N的決策為上升,劑量N+1的決策為下降,則建議採用保守的策略,暫停N+1的入組,只開展劑量N的試驗,直到可以上升了,再繼續N+1的試驗。
參考文獻
Liu, S. & Yuan, Y. (2015) Bayesian optimal interval designs for phase I clinical trials. Applied Statistics, 64, 507–523.
Zhou, H., Yuan, Y., Nie, L. (2018). Accuracy, Safety, and Reliability of Novel Phase I Trial Designs. Clinical Cancer Research, 24, 18, 4357-4364.
FDA.Drug Development Tools: Fit-for-Purpose Initiative. December 10, 2021. https://www.fda.gov/drugs/development-approval-process-drugs/drug-development-tools-fit-purpose-initiative.
Liu,S. & Yuan, Y. (2022) Erratum: Bayesian optimal interval designs for phase I clinical trials. Applied Statistics, 00, 1-2.
完結













0則評論