
开篇声明:1:以下内容转自以下内容转自微信公众号CMAC发布《夏结来教授谈“CFDI临床试验数据管理和统计分析核查要点”,FDA看了都说稳~》,如有侵权,可联系删除
2. 以下观点,仅代表作者个人观点,请带着质疑的态度去阅读。如有问题,欢迎评论区留言或直接联系作者
夏结来教授谈“CFDI临床试验数据管理和统计分析核查要点”,FDA看了都说稳~
编者按
西安空军军医大学教授夏结来于2025年3月18-20日参加苏州国际博览中心举行的2025 CMAC年会。在“全球同步与出海落地篇:与全球同步、与未来同行”板块,“国际递交临床试验监管核查的要求与实战分享”分论坛,作为一名资深的临床研究与统计从业者,他依据已经完成的国家药品监督管理局食品药品审核查验中心(CFDI)委托研究课题,深度解析了CFDI对于临床试验数据管理和统计核查要点,先培训,再规范,后核查。这里略作修改后,加以发表。
以下为夏结来教授演讲的文章全文:
一 课题概况
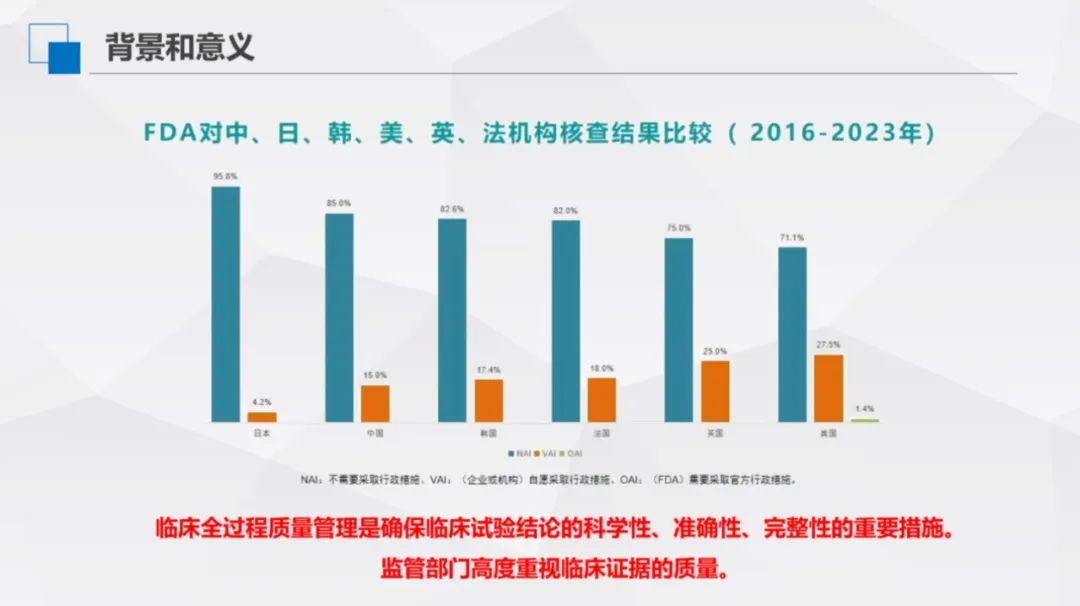
图1
伴随国内监管工作推进及行业对临床试验认知加深,中国临床试验数据质量显著提升,逐步向国际先进医药研发国家靠拢,既彰显临床试验管理的积极成果,也再次印证强化临床质量管理、筑牢数据可靠性的重要意义。
2016-2023 年,FDA对中、日、韩、美、英、法等国机构展开核查,核查结果体现了全过程质量管理在临床试验中的关键作用,反映出监管部门对临床证据质量的高度重视,也使得临床试验结论科学性、准确性、完整性得到了空前的提升。核查结果分为三类,即NAI:不需要采取行政措施;VAI:(企业或者机构)自愿采取行政措施;OAI:(FDA)需要采取官方行政措施,仅美国占 1.4%,其余国家该时段无记录。
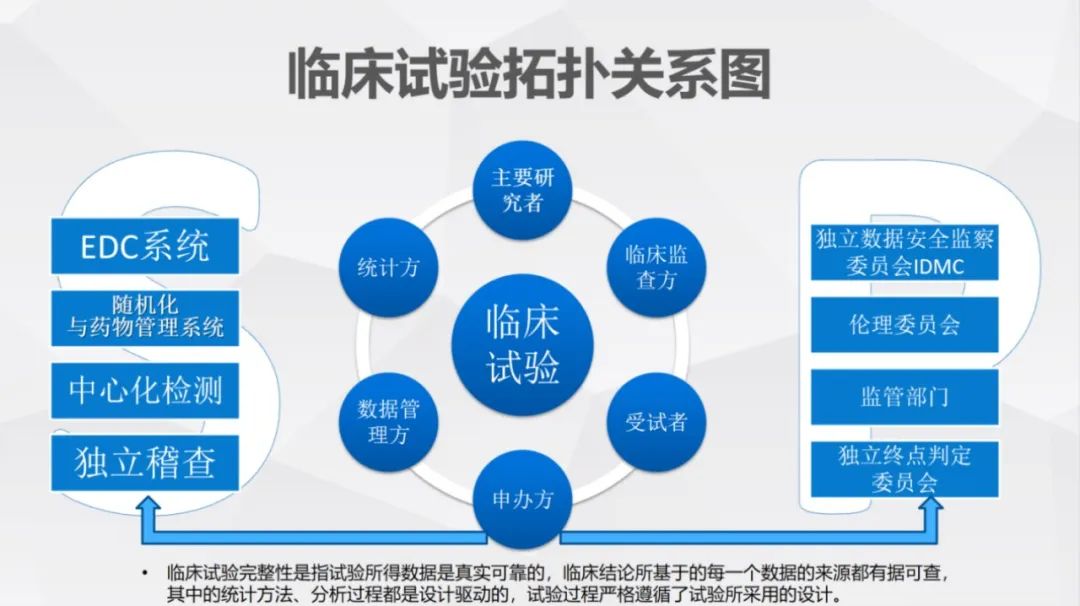
图2
其中,临床试验完整性是指试验所得数据是真实可靠的,临床结论所基于的每一个数据的来源都有据可查,其中的统计方法、分析过程都是设计驱动的,试验过程严格遵循了试验所采用的设计。需要以临床试验为核心的主要研究者、临床监查方、受试者、申办方、数据管理方、统计方,共同进行精细管理,既确保数据真实,又规范流程。
在此背景之下,为了确保真实性,CFDI承担了大量的基于不同任务目的的现场核查事务。为了进一步提高核查效力,做到有的放矢,进一步规范指导核查行动,有必要对临床试验的不同环节制定相应的核查要点和核查手册。为了满足临床试验质量评价与数据管理和统计分析具体环节核查的新需求,我们承接了CFDI指令性课题。课题组成立两个研究小组,从数据管理和统计分析两部分分别进行研究,围绕完整性与科学性两方面要求针对重点环节一一形成核查要点和关注内容。经过两年多的努力,《药物临床试验数据管理和统计分析检查员手册》已编撰完成,并以结题报告的形式提交了CFDI。
二 课题开展情况、研究结果
1.数据管理组
图3
数据管理课题小组开展了1次线下分工和框架讨论,并通过6次线上讨论和两轮意见征集,明确了质量评价和核查的要点,并建立了系统的目录结构。确定了一级目录4条,涵盖准备阶段、进行阶段、结束阶段、数据管理体系-延伸核查;二级目录14条,进一步细化各阶段的核查内容;三级核查要点122条,均明确了相应的风险等级。
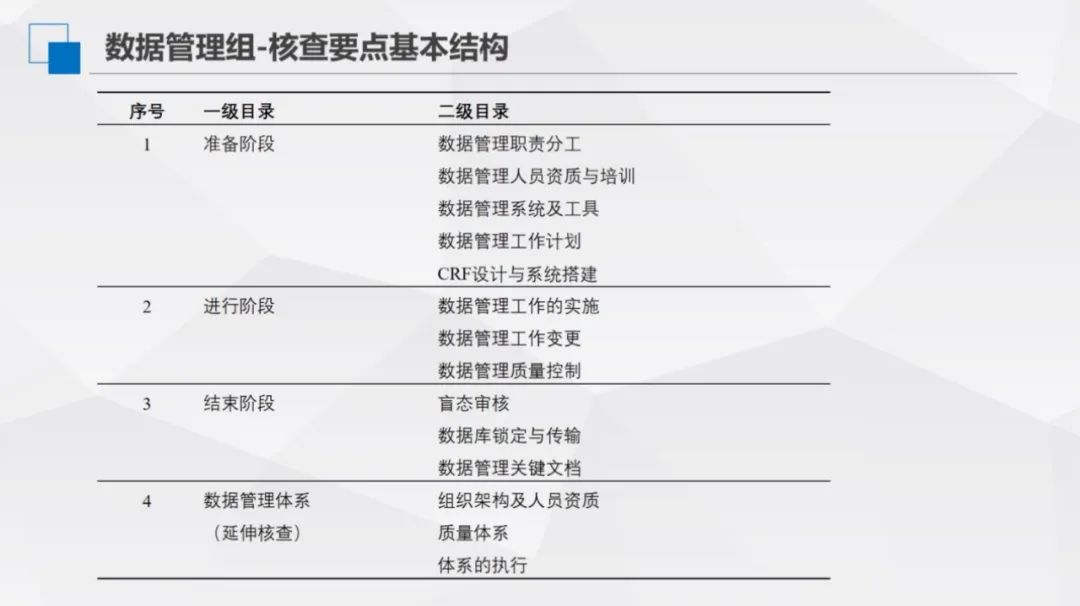
图4
(1)准备阶段:明确数据管理职责分工,核查人员资质证明、培训记录,确认数据管理系统及工具合法使用,审查数据管理工作计划制定情况,以及CRF设计与系统搭建的规范性,为数据管理奠定基础。
(2)进行阶段:聚焦数据管理工作实施,包括工作变更记录与质量控制。核查实施流程规范性、变更合理性及质量控制有效性,确保数据管理过程符合要求。
(3)结束阶段:开展盲态审核,要求留存审核过程记录,规范数据库锁定与传输操作,核查数据管理关键文档完整性。记录是工作执行的重要证明,无记录则难以佐证工作落实。
(4)数据管理体系(延伸核查):延伸核查组织架构、人员资质、质量体系建设及执行情况。若前期核查发现问题,将深入审查质量管理体系,判断其是否流于形式。若在此阶段核查出现问题,直接与项目管理体系相关联。此举不仅是核查手段,更旨在推动建立高质量数据管理队伍,明晰岗位责任,从体系层面保障数据管理规范性,避免因体系漏洞引发对整体数据管理的质疑,实现“以查促建”,提升数据管理最终成果质量。
本研究共制定了4个基本维度,共计122个核查要点。根据临床试验数据管理中的风险点、后果严重程度及现场核查的可操作性,指定了15个关键要点三级目录。
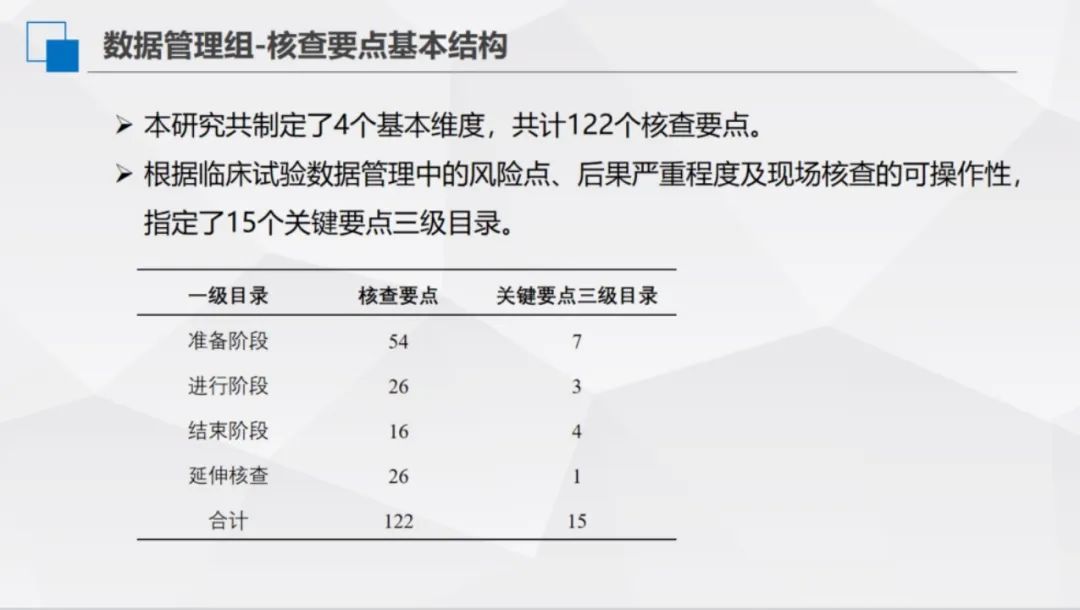
图5
2.统计分析组
图6
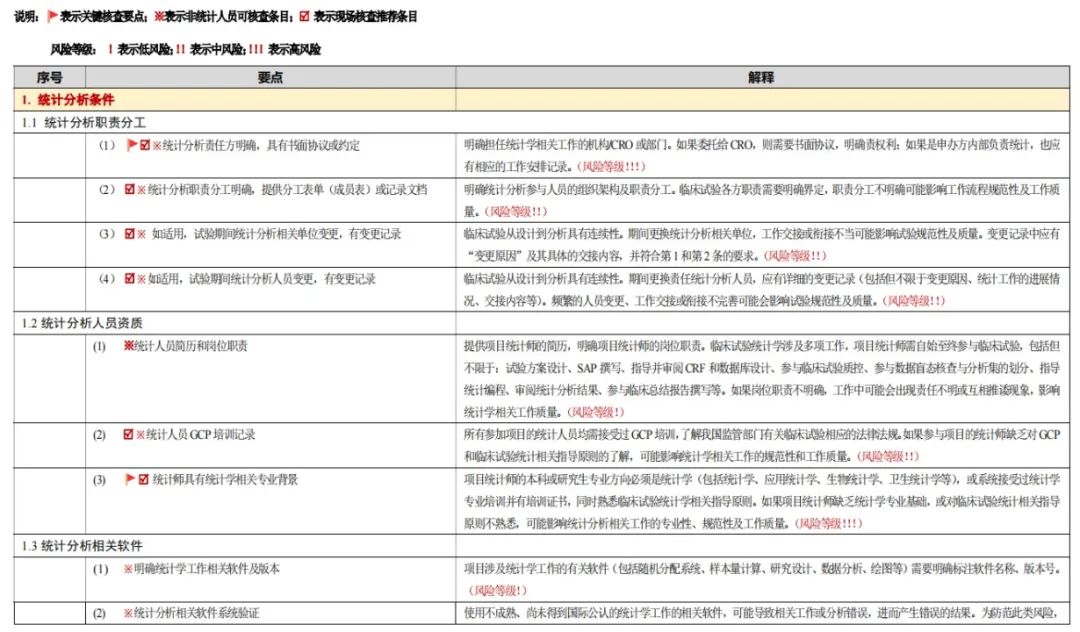
统计分析课题小组采用线上线下结合的工作模式推进研究。线上通过6次讨论,确定一级目录6条(含统计分析条件、统计学设计、统计学执行、统计分析必备文档、特殊核查要点、延伸核查)、二级目录25条、三级目录123条,明确技术要点与现场核查要点,保留不适用于现场核查的统计专属考量部分,并面向泰格、复星、康特瑞科、齐鲁等企业内部统计师收集首轮意见。
线下于2023年9月24日召开统计核查要点定稿会,经讨论征集意见,最终界定关键核查要点14条、现场核查推荐条目82条、非统计人员可核查条目88条,划分风险等级(低、中、高风险)。2023年10月针对123条核查要点形成解释文档,经意见征求,于12月10日小组总结会正式定稿。该模式融合线上讨论的高效性与线下会议的深入性,确保统计分析核查体系的科学性、规范性,推动课题工作有序完成。

图7
(1)统计分析条件:核查职责分工、人员资质及相关软件使用;
(2)统计学设计:涵盖估计目标、Ⅰ/Ⅱ类错误控制、样本量估计、分析方法、敏感性分析的设计;
(3)统计学执行:涉及SAP制定、随机化和盲法、分析人群划分、编程及结果报告等环节;
(4)统计分析必备文档:包含分析相关文档、随机化盲法文档IDMC文档、特殊文档等;
(5)特殊核查要点:针对开放试验、单臂设计、期中分析、适应性设计等特殊场景;
(6)延伸核查:关注SOP及版本控制、人员管理、系统验证等延伸内容,确保统计分析全流程规范。
本研究共制定了6个基本维度,共计122个核查要点。根据临床试验统计分析中的风险点、后果严重程度及现场核查的可操作性,指定了36条关键要点和83条现场核查推荐条目。
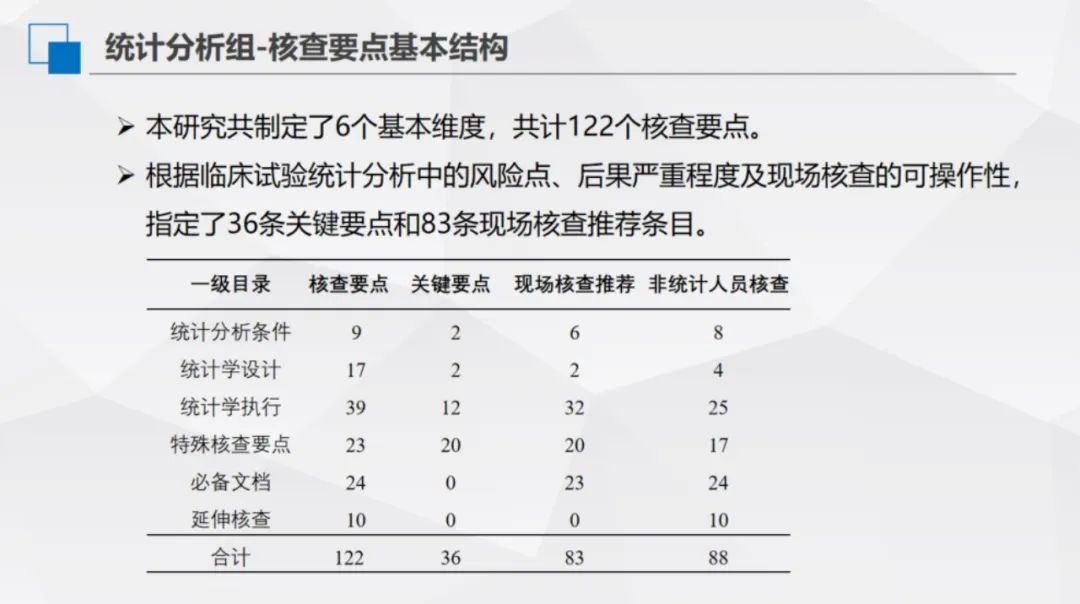
图8
统计分析组核查侧重点围绕五大共性问题展开。
第一,SAP制定。需由试验统计学专业人员起草,确保与统计分析内容一致,重点关注数据驱动的计划变更。
第二,随机化及盲法。评估执行不规范引发的试验结果偏倚,关注变更7对试验完整性、Ⅰ类错误率、统计分析及结果解读等各方面的影响。
第三,分析人群划分。强调统计师参与,关注数据驱动的划分导致假阳性错误率的增加。
第四,结果报告。强调结果的可重现,与统计分析计划的一致,避免有选择性的报告,关注不恰当的事后修改或增删引入偏倚。
第五,IDMC。强化期中分析规范执行,关注不规范的操作对试验完整性和科学性的破坏。
三 总结与展望、成果展示
1.总结与展望
本研究通过专家座谈、分组讨论、意见征集等形式,制定了适用于我国药物临床试验数据管理和统计分析工作的现场核查要点并形成检查员手册,既具有科学性,又满足合规性要求。
从内容上看,既包括现场核查需要检查的内容,又规定进一步对质量体系的核查内容。故本核查要点既能用于指导核查员开展现场核查工作的要求,又可用于申办方、合同研究机构、研究者自查,以便进一步提高临床研究的质量,并不断改进自身的质量管理体系。
未来将根据实际工作中的反馈,对本要点作进一步的完善;同时课题组将积极与行业协会合作,将本要点打造成行业标准,以期不断提高我国药物临床试验数据管理和统计分析工作的水平和质量,形成监管部门、学术界及工业界合作的范式。
2.成果展示
数据管理质量评价检查员手册(V1.0)
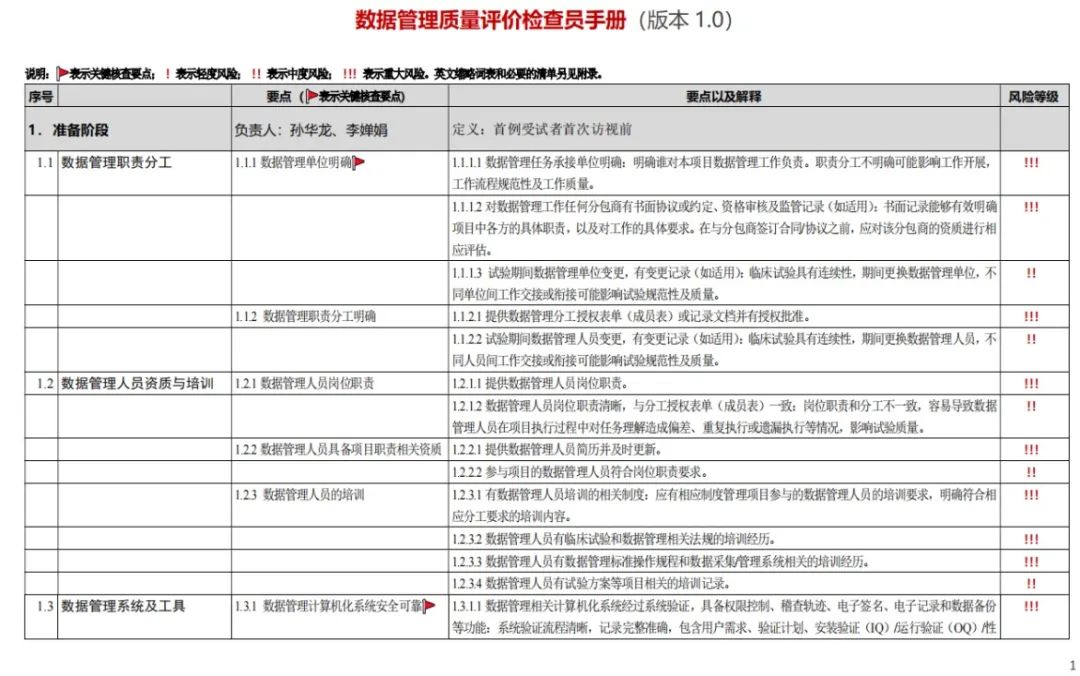
统计分析质量评价检查员手册(V1.0)
作者简介

博士生导师,国家药品管理局资深统计学审评咨询专家。
现任CSCO生物统计学专家委员会主任委员、历任中国信息协会统计理论与方法专业委员会副主任委员、中华预防医学会生物统计学分会侯任主任委员、中国卫生统计杂志编委、中国临床试验统计学组副组长、中国临床试验数据管理学组组长。
先后获8项国家自然科学基金资助,与国家药品审评中心联合申报十三五重大科技专项1项。获国家科技进步二等奖2项、军队科技进步一、二等奖各1项。提出了回归系数根方估计和广义根方估计新方法。
先后在在Statistics in Medicine, The New England Journal of Medicine等SCI收录的期刊上以共同第一作者或通讯作者发表论文40余篇。
设计:李阳
编辑:李杰
审核:夏结来
本文整理自:【中国(苏州)创新药物医学大会暨2025CMAC年会“国际递交临床试验监管核查的要求与实战分享”分论坛】
完结















0条评论