
開篇宣告:1:以下內容轉自以下內容轉自微信公眾號 阮博臨藥微研《FDA臨床藥理評審報告解讀-烏帕替尼(上)》,如有侵權,可聯絡刪除
2. 以下觀點,僅代表作者個人觀點,請帶著質疑的態度去閱讀。如有問題,歡迎評論區留言或直接聯絡作者
閱讀FDA的新藥臨床藥理報告,是每一個藥物研發人的必修課。今天開始我們定期來跟大家一起解讀NDA資料裡的臨床藥理報告。這一期選的藥物是烏帕替尼(Upadacitinib),作為艾伯維公司研發的高選擇性JAK1抑制劑,已在全球多個市場獲批用於治療類風溼關節炎(RA)及其他免疫性疾病。本文基於FDA的NDA報告中的CLINICAL PHARMACOLOGY AND BIOPHARMACEUTICS REVIEW部分,解讀烏帕替尼從Ⅰ期到Ⅲ期的臨床試驗設計邏輯。
一、總計30項臨床研究的整體佈局
烏帕替尼的臨床開發計劃由22項Ⅰ期研究、2項Ⅱ期研究(外加1項在日本開展的Ⅱb/3期支援性研究)和5項Ⅲ期研究構成,同時提交了8項體外研究和3項定量藥理學分析報告。
從研究型別分佈可以看出,Ⅰ期研究佔據了主體地位(22/30),涵蓋了PK/BA研究、內在因素影響、外在因素影響、ADME等全方位探索。這種"厚積薄發"的研究策略,確保了後續Ⅱ/Ⅲ期研究的穩健推進。
二、緩釋製劑的開發策略:從IR到ER的精準橋接
烏帕替尼的製劑開發經歷了一次重要的轉型:從速釋(IR)膠囊轉向緩釋(ER)片劑。這一轉變並非一蹴而就,而是透過一系列精心設計的相對生物利用度研究逐步推進,最終確立了每日一次的給藥方案,併爲上市制劑的生物等效性提供了直接證據。
1 核心製劑橋接研究:M14-680的系統性探索
M14-680研究是製劑轉換的核心證據之一。該研究設計了六個部分,系統比較了不同製劑和給藥方案下的藥代動力學(PK)特徵。
● Part 1 & 2(單次給藥):比較了15 mg/30 mg ER片與12 mg/24 mg IR膠囊的相對生物利用度(BA)。
√ 主要結果: 在空腹條件下,單次給予15 mg和30 mg ER片劑相較於同等總劑量的IR膠囊,AUC(暴露量)相當,但Cmax(峰濃度)降低了約63%。
這表明ER製劑能有效平滑血藥濃度峰值。此外,高脂高熱飲食使30 mg ER片劑的Cmax和AUC分別增加了20%和17%,提示食物影響較小且無臨床意義。
● Part 3(多次給藥ER):評估ER片劑多次給藥的PK。
√ 主要結果: 每日一次(QD)給藥後,穩態在4天內達到,且蓄積極少。15 mg和30 mg QD劑量範圍內的PK呈線性比例。
● Part 5 & 6(多次給藥橋接):比較15 mg ER QD vs 6 mg IR BID,以及30 mg ER QD vs 12 mg IR BID。
√ 主要結果:多次給藥達穩態後,15 mg ER QD與6 mg IR BID的全身暴露量(AUC)相當;30 mg ER QD與12 mg IR BID的全身暴露量(AUC)相當。
這直接證實了ER製劑可以實現每日一次給藥,同時維持與有效II期劑量(IR BID)相似的暴露水平,為III期臨床選擇QD方案奠定了堅實基礎。
2 上市前橋接與特殊劑量探索
在確立ER製劑為核心開發方向後,研發團隊進一步開展了兩項關鍵的補充研究:
● M15-878研究(上市影像製劑橋接):比較了“上市影像製劑”(To-be-marketed, ER17/ER18)與Ⅲ期臨床所用製劑(ER7/ER8)的BA。
√ 主要結果:研究顯示,15 mg和30 mg規格的上市制劑與Ⅲ期臨床製劑在空腹條件下生物等效(Bioequivalent)(Cmax和AUC的90%置信區間均在80-125%範圍內)。
高脂飲食使30 mg上市制劑的Cmax增加40%,AUC增加30%,但這被認為無臨床相關性。這一結果確保了商業化批次與臨床批次的療效和安全性一致。
● M16-094研究(高劑量探索):評估了45 mg單一片劑與30 mg+15 mg聯合用藥的相對BA及食物影響。
√ 主要結果:空腹條件下,單片45 mg ER片劑與15 mg+30 mg兩片聯用的AUC和Cmax相當。高脂飲食使Cmax和AUC分別增加18%和30%。該研究為未來可能的高劑量適應症拓展儲備了關鍵資料。
3 首次人體研究:M13-401的探索性價值
M13-401作為首次人體試驗(FIH),承擔了探索劑量範圍和建立安全性耐受性基線的重要使命。
√ 主要結果:
劑量遞增(SAD):明確了烏帕替尼在健康志願者中的最大耐受劑量,且Cmax和AUC在1-48 mg範圍內呈劑量比例關係。
食物影響:與空腹相比,非空腹條件下給藥導致Cmax降低約23%(注:此結果為IR製劑早期資料,後續ER製劑受食物影響表現爲輕度增加,但均無臨床意義)。
DDI(酮康唑):與強CYP3A4抑制劑酮康唑合用,烏帕替尼的Cmax、AUC0-t和AUCinf分別增加了約70%、76%和75%。這一發現直接導致了說明書中關於“慎與強CYP3A4抑制劑合用”的建議。
4 其他製劑探索:完善證據鏈的補充研究
● M14-678研究(低劑量ER):比較7.5 mg ER片與2×3 mg IR膠囊的相對BA。
√ 主要結果:7.5 mg ER片相對於3 mg IR BID的相對生物利用度約為66%-86%。該研究為日本等地的低劑量(7.5 mg)適應症開發提供了資料支援。
● M16-552研究(口服溶液):評估口服溶液製劑的BA。
√ 主要結果:空腹條件下,分兩次給予6 mg口服溶液(間隔12小時)相較於單次15 mg ER片劑,Cmax高出30%,AUC高出17%。
該資料為兒童用藥或吞嚥困難患者的未來製劑開發提供了參考,表明溶液劑型可能需要調整劑量以匹配片劑暴露量。
科學意義小結:
透過從首次人體研究(M13-401)到核心製劑橋接(M14-680),再到上市前驗證(M15-878)和特殊場景儲備(M16-094、M14-678、M16-552)的完整研究鏈條,烏帕替尼的製劑開發完成了從IR到ER的轉型。
關鍵資料證實15 mg ER QD與6 mg IR BID暴露量相當,30 mg ER QD與12 mg IR BID暴露量相當,且上市制劑與臨床製劑生物等效,為NDA申報和後續市場供應奠定了堅實的科學基礎。
三、暴露-效應分析:15 mg vs 30 mg的劑量抉擇
在劑量選擇這一關鍵決策點上,烏帕替尼的研發團隊展現了定量藥理學的強大力量,整合了Ⅱ期(M13-537, M13-550)和Ⅲ期研究資料。
1 暴露-療效關係:平臺期的識別
研究團隊採用連續時間馬爾可夫模型對ACR20/50/70應答率等療效終點進行建模。
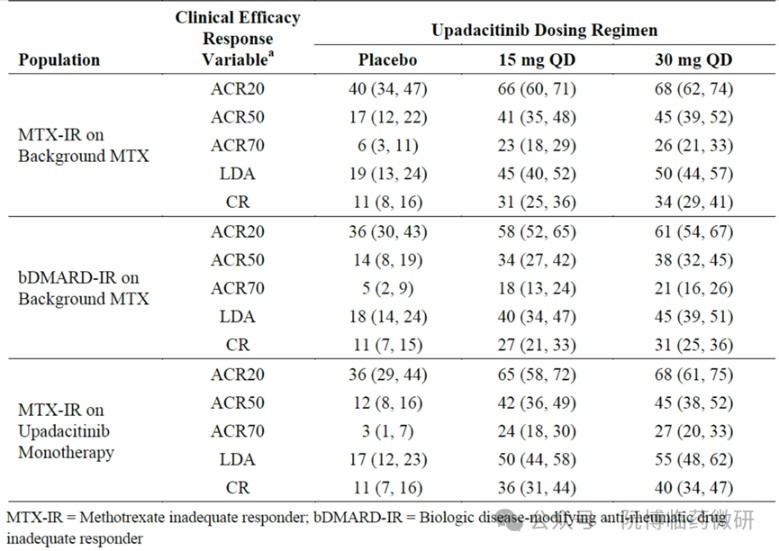
√ 主要結果:模擬結果顯示,30 mg QD劑量相較於15 mg QD僅能提供微小的增量療效獲益。例如,在ACR20響應率上,30 mg組雖有數值上的升高趨勢,但與15 mg組相比無具有臨床意義的顯著提升。這表明15 mg QD已達到療效平臺期,更高劑量並不能帶來有臨床意義的療效提升。
2 暴露-安全性關係:劑量限制性毒性的識別
安全性分析採用了分層遞進的策略,重點考察了血紅蛋白、感染等指標。
√ 主要結果:
血紅蛋白下降: 血紅蛋白下降幅度(>1 g/dL和>2 g/dL)的發生率與藥物暴露量呈正相關,30 mg QD組的風險顯著高於15 mg QD組。
嚴重感染: 截至24/26周,嚴重感染的發生率與暴露量存在關聯,高暴露量(對應30 mg劑量)組發生率更高。
其他指標: 肺炎、帶狀皰疹、血小板變化等未顯示明確的暴露-反應關係。
√ 決策結論:
綜合療效與安全性的暴露-效應分析,最終支援選擇15 mg QD作為推薦劑量,30 mg QD作為備選方案。
這一決策過程充分體現了"以患者為中心"的藥物開發理念——在保證療效(15 mg已達平臺期)的同時,儘可能降低劑量相關的安全風險(如貧血和嚴重感染)。
總結
透過嚴謹的暴露-效應分析,烏帕替尼的研發團隊在療效與安全性之間找到了最佳平衡點,最終確立了15 mg這一兼具療效與安全性的推薦劑量。至此,劑型與劑量兩大核心要素已獲得充分的臨床資料支援。
下一篇,我們將繼續解析研發團隊如何回答臨床應用中不可迴避的關鍵問題:
哪些藥物存在合用禁忌?
肝腎功能受損的患者是否需要調整劑量?
以及如何透過群體藥代動力學研究整合上千例患者的臨床資料,為真實世界的精準用藥提供全景式指導。
完結










Kowalevski
還是要關注後續的臨床效果
回覆dwy
@Kowalevski: 好的
回覆